How can unique molecular identifiers (UMIs) help to reduce quantitative biases?
Maybe you have already heard about UMIs and how they can help to reduce errors and quantitative biases in your Next-Generation Sequencing (NGS) experiment. But do you know how they work and how you should handle them bioinformatically after sequencing? Here we will shortly introduce these molecular tags, which are added to the DNA fragments of your sample.
Explanation
Unique molecular identifiers (UMIs), or molecular barcodes (MBC) are short DNA molecules which are ligated to your DNA fragments. The random sequence composition of the UMIs assures that every fragment-UMI combination is unique in your library. After PCR enrichment, without UMIs, one can not distinguish if multiple copies of a fragment are caused by PCR clones or if they are real biological duplicated. By using UMIs, PCR clones can be found by searching for non-unique fragment-UMI combinations, which can only be explained by PCR clones.
When performing variant analyses, these falsely overrepresented fragments can result in incorrect calls and thus wrong diagnostic findings (see Figure 1).
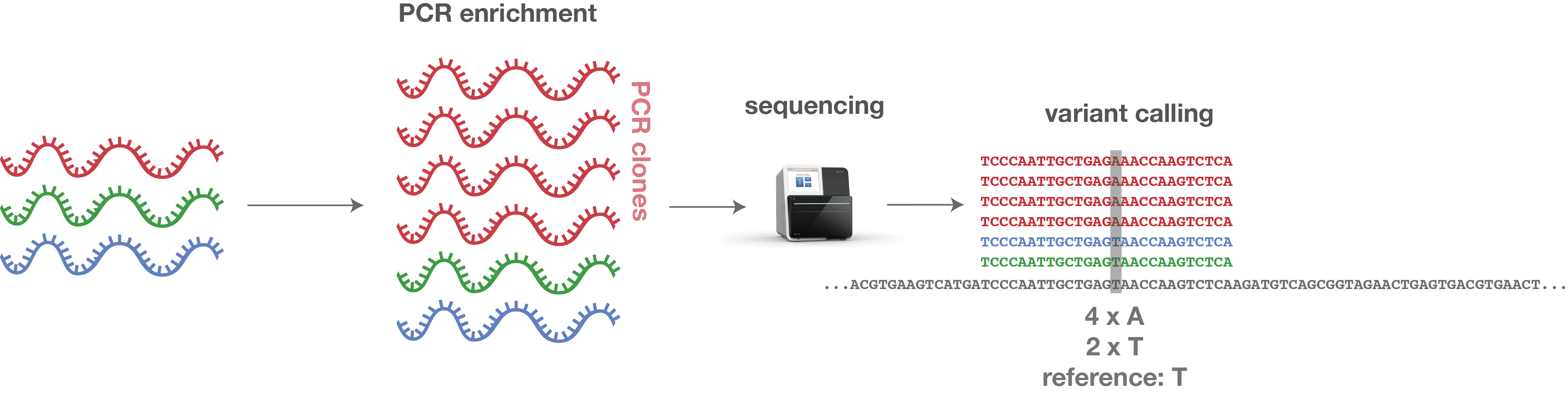 Figure 1: Error produced by PCR clones.
Figure 1: Error produced by PCR clones.
De-Duplication
The process of discarding PCR clones, ending up with one single representative is known as de-duplication. De-duplication can be performed on the raw reads, before the mapping process or after mapping (recommended). If the de-duplication is performed after mapping, the sequence of the UMIs need to be clipped away, since these synthetic nucleotide strings do not occur in the genome. After clipping, the UMI sequences have to be stored for every single read, since they are needed for de-duplication after mapping. If there are stacks of reads, all starting and ending at the exact same positions and having the same UMI sequence, PCR clones have been detected and can easily be corrected by deleting all copies, leaving one representative (see Figure 2).
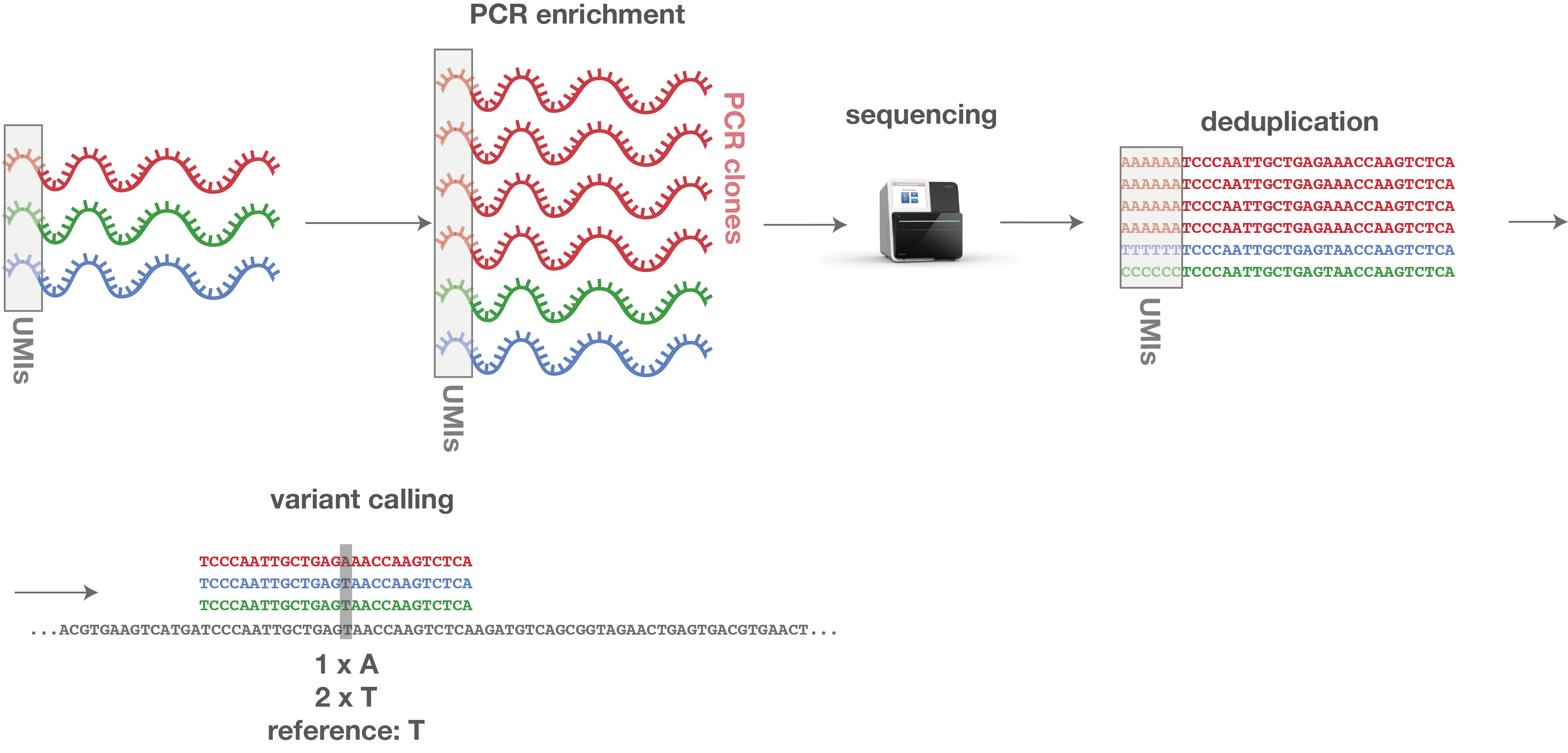 Figure 2: After de-duplication using UMIs, variants can be correctly called.
Figure 2: After de-duplication using UMIs, variants can be correctly called.
Add-On: Quality Enrichment
If the de-duplication is performed after the mapping step, one can use the multiple copies of PCR clones to enricht the quality of the read for the original fragment. Since the fragment was duplicated before sequencing and then sequenced several times, the multiple copies can be used to correct for sequencing errors. By performing a majority voting over all multiple copies (having the same UMI sequence) of every single genomic position, one can detect the original nucleotide sequence of the original fragment (see Figure 3).
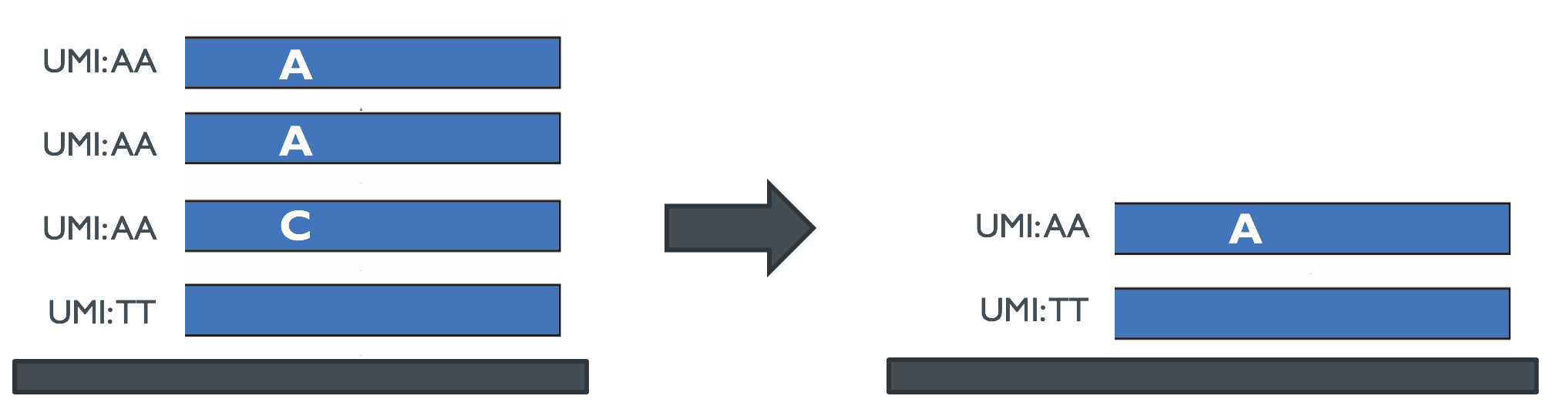 Figure 3: After mapping, duplicated sequences can be used for correcting sequencing erros.
Figure 3: After mapping, duplicated sequences can be used for correcting sequencing erros.
Conclusion
Using UMIs and thus correcting for these errors became obligatory in current diagnostic panels. Correct bioinformatics processing of libraries with UMIs is not trivial and result in strong biases, if it is done incorrectly. Unfortunately, there is still a lack of well established tools.
Promotion
The genetic testing software Seamless NGS is able to correctly clip UMIS and de-duplicate PCR artifacts after mapping.
Would you like to sharpen your NGS data analysis skills?
Join one of our public workshops!About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on March 20, 2017