How do strand specific sequencing protocols work?
NGS has become a powerful research tool for studying biological macromolecules such as RNA. Before sequencing, RNA samples have to be prepared using a protocol where the single-stranded RNA molecules are first converted into double-stranded complementary DNAs (cDNAs), followed by adapter ligation and PCR amplification. After sequencing, the generated reads can be mapped back to their transcripts of origin. For each of the transcripts, by tabulating the number of mapped reads and by normalizing read counts, it is possible to estimate the expression level of transcripts and genes.
The problem with non-strand-specific protocols
At the beginning, most sample preparation protocols are non-strand-specific (NSS). This means that sequences of both sense strand and the antisense strand of the original mRNA are obtained, without knowing which strand of the cDNA corresponds to the original mRNA. Consequently, it is not clear if a read originated from the sense strand or the antisense strand of the reverse transcripted mRNA.
Ideally, you would like to assign all reads coming from molecules of the sense strand to a sense strand fragment/genome/reference and all reads coming from molecules of the antisense strand to an antisense fragment/genome/reference. Unfortunately such an assignment is not possible by using non-strand-specific protocols. It is like having two puzzles where some pieces of the one puzzle also fit with the other one and you cannot assign which belongs to which. That is why in some cases you will map some of the antisense strand reads to the sense fragment/genome/reference or the other way around as shown in Fig. 1A. This possibly wrong assignment can lead to biased estimates of transcription, especially for transcripts overlapped at the antisense strand (see following section).
 Figure 1: Comparison of mapped reads from an unstranded/stranded library.
Figure 1: Comparison of mapped reads from an unstranded/stranded library.
In this context it is important to know that there are also some (regulatory) genes on the antisense DNA strand (Fig. 2). An example for such overlapping transcripts are cis-natural antisense transcripts (NATs). These transcripts are coded on the antisense strand overlapping with a transcript on the opposite sense strand. Antisense transcription is a mechanism for gene regulation and gene silencing that has been reported to be widespread in the mammalian transcriptome [3,4]. The resulting NAT-RNA is a part of the sense strand but can also map to the antisense strand because of the overlapping base sequence.
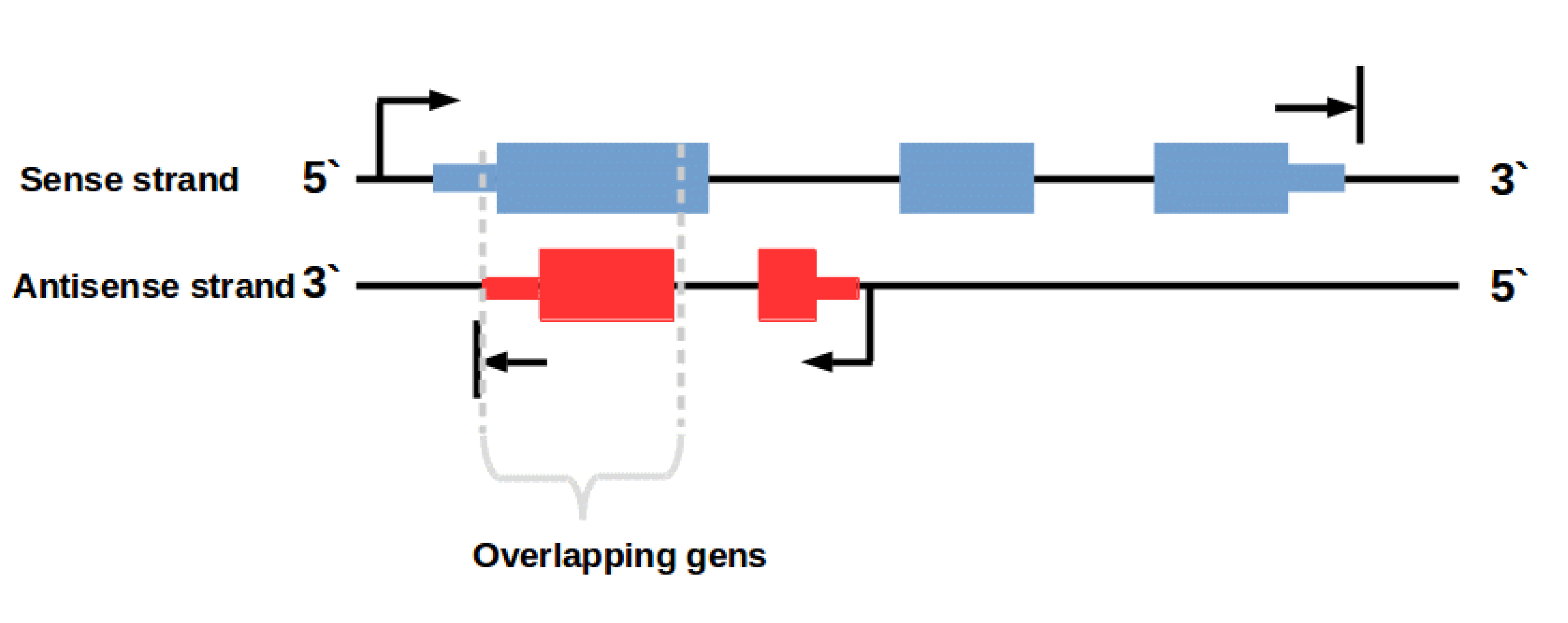 Figure 2: Overlapping genes on sense and antisense strand. Transcription of genes on the antisense strand creates regulatory RNAs e. g. cis-natural antisense transcripts (NATs)
Figure 2: Overlapping genes on sense and antisense strand. Transcription of genes on the antisense strand creates regulatory RNAs e. g. cis-natural antisense transcripts (NATs)
Available strand-specific protocols
Strand-specific protocols have been developed to overcome these ambiguities. Their aim is to allow an assignment of the reads to their original strand. In general this can be managed in two different ways: One strategy is based on attaching different adapters in a known orientation relative to the 5’ and 3’ ends of the RNA transcript. The subsequent reverse transcription and amplification step creates a cDNA library flanked by two distinct adapter sequences, whereby the orientation of the adapters for the original mRNA is known. From now on it is possible to assign the produced reads for read mapping (Fig. 1B). This strategy exists in various variants [5]. For Illumina systems the most widely used variant is the on‑flowcell reverse transcription (FRT-seq) [6], shown in Fig. 3.
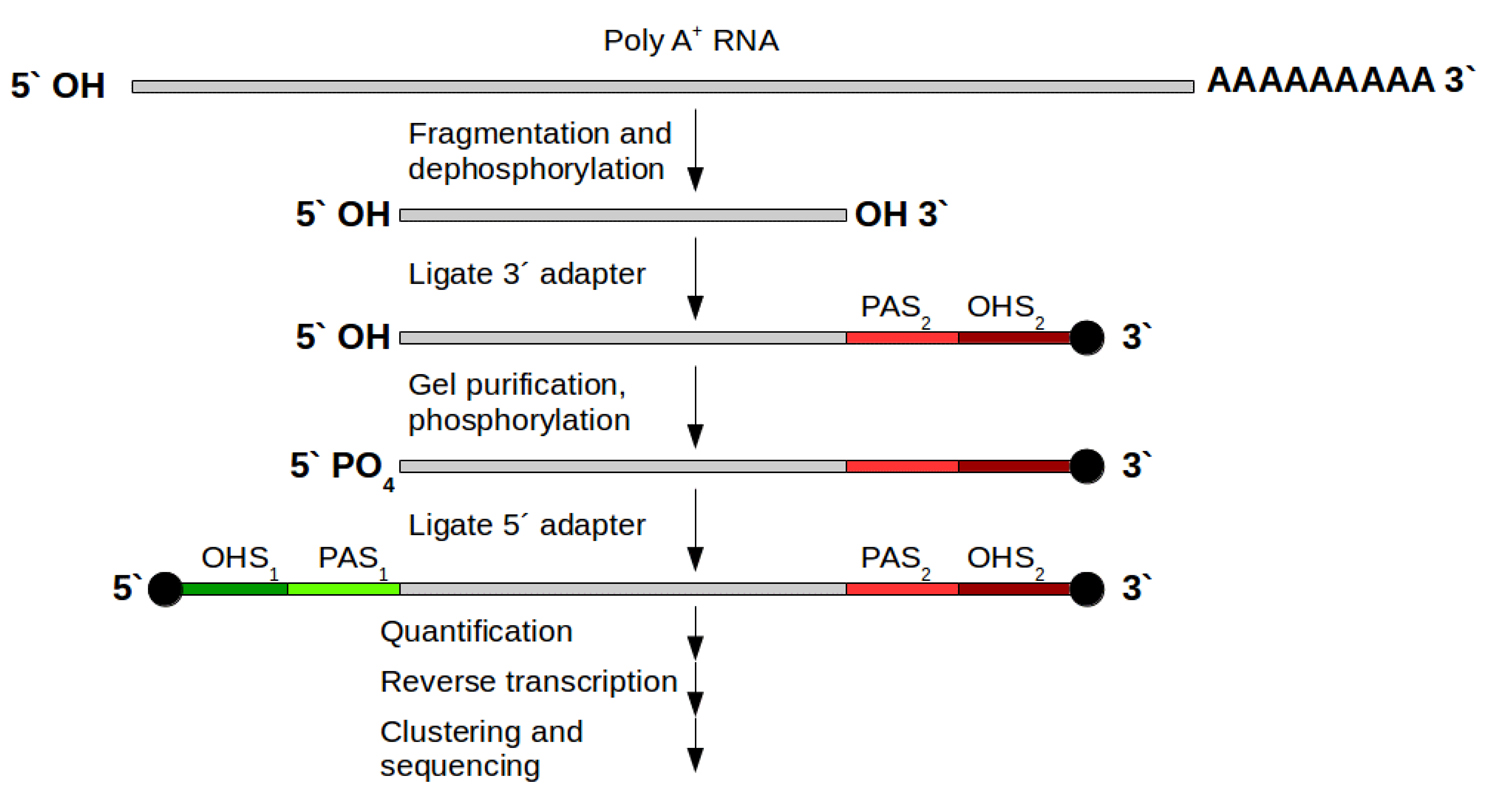 Figure 3: FRT-seq workflow for attaching different adapters on mRNA to keep direction information. Grey: poly‑A selected mRNA; red and green parts: distinct attached adapters including 2 regions per adapter: PAS (primer attachment site) and OHS (oligo hybridization site) for following hybridization on the flow cell and bridge amplification. After this, one orientation is removed and sequencing can start for the remaining DNA fragments.
Figure 3: FRT-seq workflow for attaching different adapters on mRNA to keep direction information. Grey: poly‑A selected mRNA; red and green parts: distinct attached adapters including 2 regions per adapter: PAS (primer attachment site) and OHS (oligo hybridization site) for following hybridization on the flow cell and bridge amplification. After this, one orientation is removed and sequencing can start for the remaining DNA fragments.
A second way is to mark one strand by chemical modification. It is possible to mark the RNA itself by bisulfite treatment or during second-strand cDNA synthesis followed by degradation of the unmarked strand [5].
Summary
Strand-specific protocols can be more challenging and time consuming to prepare than standard protocols for RNA-seq. But the additional information should not be underestimated and allows you to resolve otherwise ambiguous data.
References:
- Metzker, M. L. Sequencing technologies - the next generation. Nat. Rev. Genet. 11, 31–46 (2010).
- Tsai, K.-W. et al. Evaluation and Application of the Strand-Specific Protocol for Next-Generation Sequencing. BioMed Res. Int. 2015, e182389 (2015).
- Carninci, P. et al. The transcriptional landscape of the mammalian genome. Science 309, 1559–1563 (2005).
- Katayama, S. et al. Antisense transcription in the mammalian transcriptome. Science 309, 1564–1566 (2005).
- Levin, J. Z. et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 7, 709–715 (2010).
- Mamanova, L. & Turner, D. J. Low-bias, strand-specific transcriptome Illumina sequencing by on-flowcell reverse transcription (FRT-seq). Nat. Protoc. 6, 1736–1747 (2011).
Would you like to sharpen your NGS data analysis skills?
Join one of our public workshops!About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on January 11, 2018