Trimming adapter sequences - is it necessary?
Removal of adapter sequences in a process called read trimming, or clipping, is one of the first steps in analyzing NGS data. With more than 30 published adapter trimming tools there is a more than large choice for the appropriate tool. Yet, there is a debate whether this step really is as important as the number of tools suggests, or whether it is possible to skip this time-consuming step for many NGS applications.
Why do adapters contaminate my sequences?
Adapters have to be ligated to every single DNA molecule during library preparation. For Illumina short read sequencing, the corresponding protocols involve (in most cases) a DNA fragmentation step, followed by the ligation of certain oligonucleotides to the 5’ and 3’ ends. These 5' and 3' adapter sequences have important functions in Illumina sequencing, since they hold barcoding sequences, forward/reverse primers (for paired-end sequencing) and the important binding sequences for immobilizing the fragments to the flowcell and allowing bridge-amplification.
When are adapters sequences observed in the reads?
In common short read sequencing, the DNA insert (original molecule to be sequenced) is downstream from the read primer, meaning that the 5' adapters will not appear in the sequenced read. But, if the fragment is shorter than the number of bases sequenced, one will sequence into the 3' adapter. To make it clear: In Illumina sequencing, adapter sequences will only occur at the 3' end of the read and only if the DNA insert is shorter than the number of sequencing cycles (see picture below)!
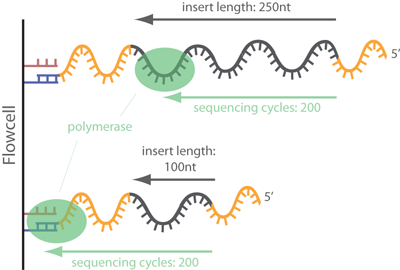
How often that happens largely depends on the used NGS protocol. Think about it: How often will you sequence into the 3' adapters when performing common RNA-Seq? After mRNA enrichment, cDNA creation (using a reverse transcriptase) and DNA fragmentation the protocols typically involve a size selection. When using a miSeq with 2x300 paired-end mode, one will select molecules that are longer than the read length, in our example greater than 600 nucleotides in length. However, it is technically impossible to obtain a specific fragment size, but one will rather get a distribution of fragment lengths (see picture). Thus, one will also obtain a certain fraction of adapter contamination for large fragment sizes. For RNA-Seq you will observe that only 0.2 - 2% of reads contain adapter sequences.
Summary
Adapter contamination will lead to NGS alignment errors and an increased number of unaligned reads, since the adapter sequences are synthetic and do not occur in the genomic sequence. There are applications (e.g. small RNA sequencing) where adapter trimming is highly necessary. With a fragment size of around 24 nucleotides, one will definitely sequence into the 3' adapter. But there are also applications (transcriptome sequencing, whole genome sequencing, etc.) where adapter contamination can be expected to be so small (due to an appropriate size selection) that one could consider to skip the adapter removal and thereby save time and efforts.
Update
As mentioned at the beginning, there is a debate about this topic. Based on this text a lively discussion started at the NGS discussion forum 'Biostars'. If you want to get more detailed information from researchers, software developers or experts in the field, we highly encourage you to visit this page and/or join this interesting discussion. There are many pros and cons that will help you making your own, thought-out decision.
Link to Biostars.org
Would you like to sharpen your NGS data analysis skills?
Join one of our public workshops!About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on August 07, 2016