What is a good sequencing depth for bulk RNA-Seq?
One of the first considerations for planning an RNA sequencing (RNA-Seq) experiment is the choosing the optimal sequencing depth. As described in our article on NGS coverage calculation, the term sequencing depth describes the total number of reads obtained from a high-throughput sequencing run. It is usually specified on a per-sample basis in the unit “numbers of reads (in millions)”. The related term sequencing coverage typically is not used in the scope of RNA-Seq: coverage represents the ratio between the number of bases of the mapped reads in relation to the number of bases of a reference in terms of redundancy (Fig. 1).
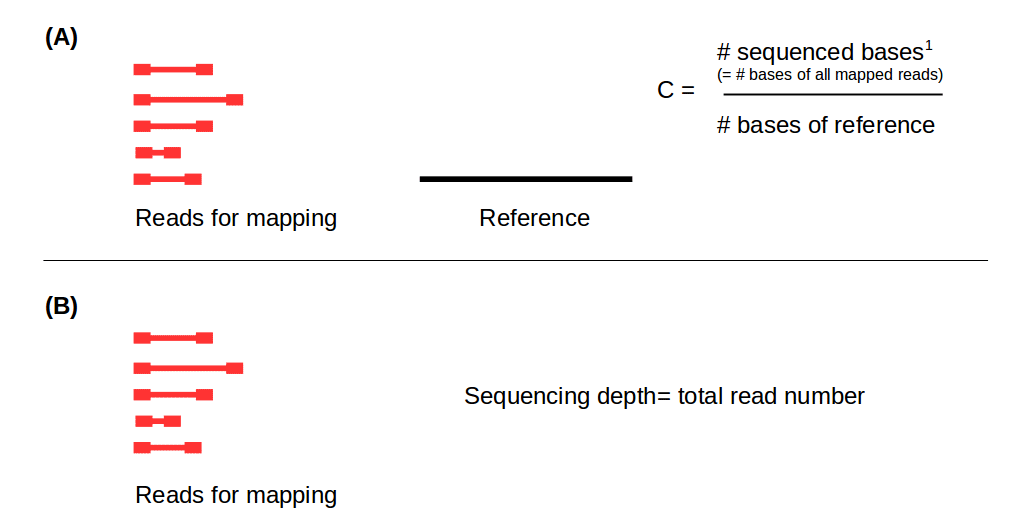 Figure 1: Distinction between coverage in terms of redundancy (A) and sequencing depth (B). C: Coverage, #: Number, 1: number of reads x read length
Figure 1: Distinction between coverage in terms of redundancy (A) and sequencing depth (B). C: Coverage, #: Number, 1: number of reads x read length
RNA-Seq sequencing depth positively correlates with information content and reliability of the obtained data: the higher the number of reads, the higher the statistical power of your results. At the same time, the sequencing costs will increase along with the number of reads. This leads to the question of what a sufficient sequencing depth is, striking a balance between the statistical power needed for the experiment and financial constraints.
How deep is enough in RNA-Seq?
The number of reads required for RNA-Seq will depend on how sensitive the experiment needs to be, the complexity of your organism and the project goals. In general 5 M mapped reads is a good bare minimum for a differential gene expression (DGE) analysis in human. In many cases 5 M – 15 M mapped reads are sufficient. You will be able to get a good snapshot of highly expressed genes. A higher sequencing depth generates more informational reads, which increases the statistical power to detect differential expression also among genes with lower expression levels. For that reason, many published human RNA-Seq experiments have been sequenced with a sequencing depth between 20 M - 50 M reads per sample. This gives a more global view on gene expression and also some information for alternative splicing analysis. There are also cases where significantly less (e.g. with targeted RNA-Seq) or significantly more (e.g. for transcriptome assembly) sequencing depth is required.
The power of replicates in RNA-Seq
For planning an RNA-seq experiment it is important to consider that a rising number of biological replicates (number of examined samples) also increases the statistical power of differential gene expression detection - a compromise should be made between sequencing depth and biological replication. In an DGE methodology experiment Liu et al. showed that, based on a sequence depth of 10 M reads per sample, raising the number of biological replicates from 2 to 6 results in a higher increase of gene detection and statistical power than raising the number of reads from 10 M ‑ 30 M.
Going on
An important step of any NGS experiment is the initial planning. Thinking about how your project goals match to available sequencing options (sequencing machine, read length, sequencing depth) helps you to avoid issues with the downstream analysis and to generate good and statistically powerful results without unnecessary expenditures. It is also helpful to research in the literature for similar previous experiments and to ask people for their experiences. Some research fields have community standards in place for their RNA-Seq experiments, for example look at the ENCODE project.
References:
- 1.Liu, Y., Zhou, J. & White, K. P. RNA-seq differential expression studies: more sequence or more replication? Bioinformatics 30, 301–304 (2014).
Interested in learning more about RNA-Seq?
Check out our upcoming course RNA-Seq Data Analysis Workshop (March 27-30, 2023 in Berlin)About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on June 18, 2019