What is the best NGS alignment software?
There is this question that comes up very often during our workshops: what is the best NGS read aligner? What alignment tool do you recommend to use for my next-generation sequencing experiments? Indeed this is a relevant question since read mapping often is a central step in any NGS analysis pipeline.
Why is it so important?
Traditional sequence alignment tools like BLAST are not suited for NGS. To extract meaningful information from NGS data, one needs to align millions and millions of mostly short sequences. BLAST and similar tools are way too slow for the vast amount of data produced by modern sequencing machines. The advent of these machines ignited the development of a class of new, much faster read aligners. Many of them: there are now more than 90 short read alignment programs available.
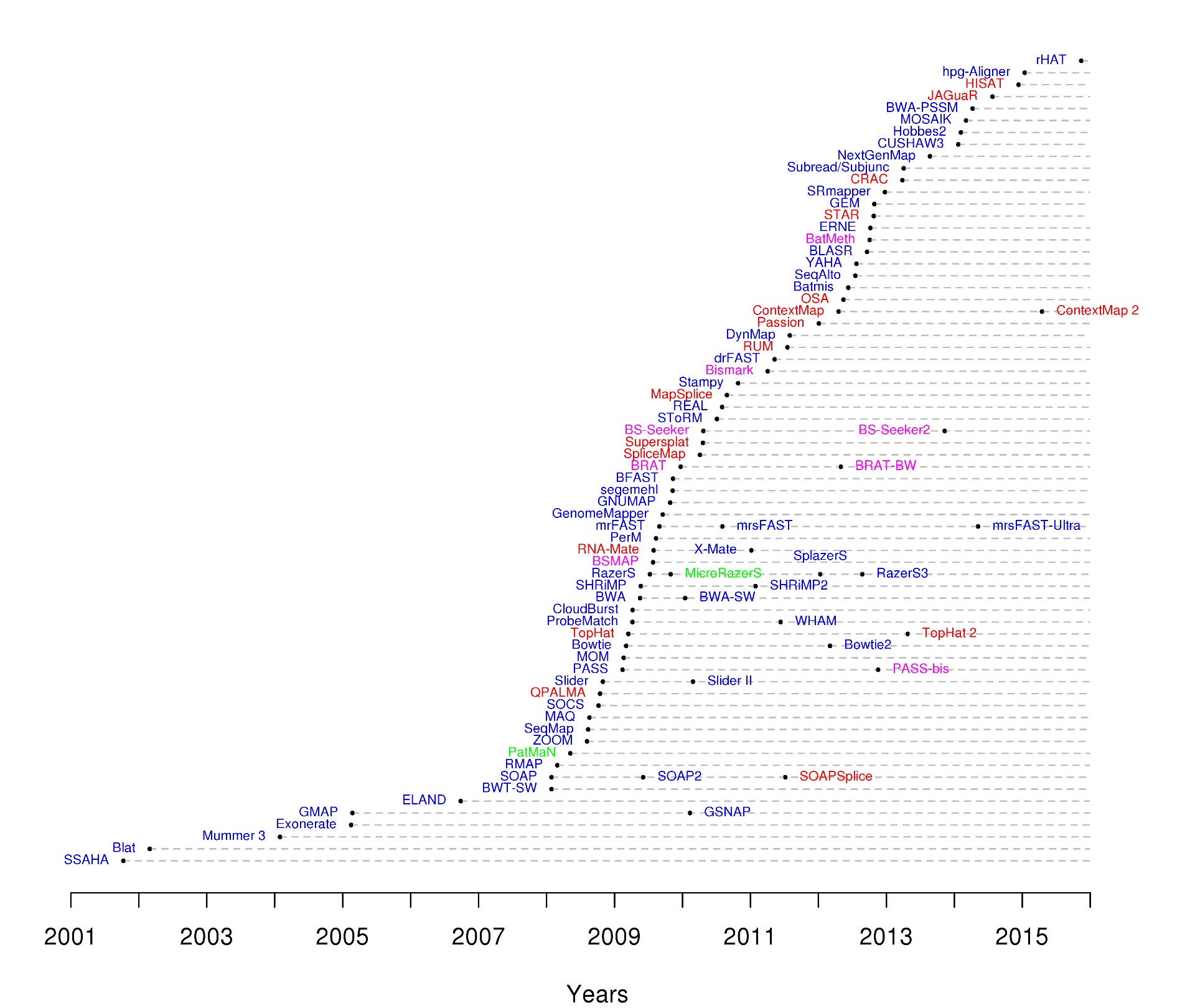 Timeline of NGS read aligners. Image from Nuno Fonseca, HTS Mappers.
Timeline of NGS read aligners. Image from Nuno Fonseca, HTS Mappers.
How to compare read alignment software?
Every time a new read alignment software is developed and officially presented in a peer reviewed journal, the authors are asked to provide a comparison to existing tools. This is typically done in a benchmark where certain aspects of a software tool are assessed (ideally) in a scientifically sound manner. You can then compare these benchmarks and use them to decide on the optimal tool for your case. However, this procedure has its limitations: only a small set of the many aspects - typically things like mapping rate, sensitivity, speed - can be assessed in a short paper. And only certain program versions, parameter settings and data can be assessed.
Click to see our comparison of read alignment software
How to decide on the best alignment software?
Assume you have a benchmark of your favorite alignment tools, what aspects should you look for? In general, you should try to answer the following questions:
- What kind of sequences/experiment do you have? Do you have fragmented DNA inserts or spliced sequences from total RNA-seq? Is there a special protocol, DNA treatment or enrichment involved? What species is it and how is the quality of its genome assembly?
- What sequencing platform do you have? Do you deal with Illumina,Ion Torrent reads or PacBio? Each machine has its characteristic read length and error types and not all mapping tools can handle them.
- What kind of further analysis do you plan to carry out with the alignments? Do subsequent tools maybe depend on the reported alignment types and formats.
- What kind of infrastructure do you have? Can you run your computations on a high performance computing cluster or does it need to run on your desktop computer?
Note that besides this “hard” benchmark there are also other factors to consider: is the output or input format of the program usable for you? Does the software have special features relevant for you? Is the program easy to use? Are there special license requirements or fees associated with it?
And the answer is...
...there is no best read aligner. It really depends on your goals and the specific case. What is the application? What sequencing technology has been used? What is the species? What are the computational constraints, etc.? You need to take into account the answer to those questions and then decide on the best read mapper according to the performance in the aspects important to you as well as in the software's features.
Would you like to sharpen your NGS data analysis skills?
Join one of our public workshops!About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on April 07, 2016