Why do Illumina reads all have the same length when sequencing differently sized fragments?
Perhaps you are surprised by the observation that you get reads of the same length when you sequence differently sized DNA fragments. No problem: you are not the first one! For NGS-beginners all the distances and sizes used during the library preparation and sequencing process can be a little bit confusing. To better understand the relationship and meaning of the terms, please take a look on the following figure.
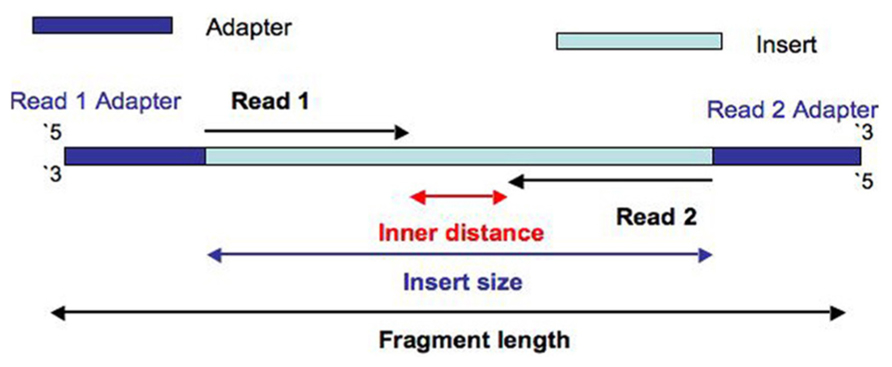 Figure 2: Basic notations of a paired end sequenced DNA fragment
Figure 2: Basic notations of a paired end sequenced DNA fragment
Basically a complete NGS fragment prepared for sequencing consists of the original DNA fragment (insert size) and the adapters added at each end. The adapters have two main functions. Firstly, they allow an orientation-specific binding of the complete fragment to the flow cell by hybridization. Secondly, they including primer binding sites needed for bridge amplification and sequencing. The adapters can also include a known sequence called unique molecular identifiers (UMIs) to be used as individual markers for each sequenced fragment.
The sequencing starts at Read 1 Adapter (mate 1) and ends with the sequencing from Read 2 Adapter (mate 2). The reads have a length of typically 50 - 300 bp. Normally the insert size is longer than the sum of the two read lengths, meaning there is an unsequenced inner part in the middle of the insert.
So why you get reads of the same length when you sequence differently sized fragments? The explanation for this is that (paired-end) sequencing always starts at the endings of the fragment, where the primer attaches, creating read 1 and (after a turnaround stage) read 2 (see Fig. 1). The length of the sequence reads then is determined by the number of sequencing cycles. The number of cycles is selected on the sequencing machine before starting the run. In each cycle, SBS extends the template strand by one nucleotide, which also extends the read length by one more base. That means that the fragment size doesn't matter at all.
We hope this article helps you to understand the relationship of fragments and NGS reads, and gives you an idea how the different parts work together during the NGS workflow.
Would you like to sharpen your NGS data analysis skills?
Join one of our public workshops!About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on May 16, 2019