MRD Tracking Software: From Molecular Signals to Longitudinal Monitoring
Clinical remission describes a state of disappearance or reduction of the signs and symptoms of disease after receiving a successful cancer treatment. In hematological diseases such as acute myeloid leukemia, this may mean that conventional diagnostic methods no longer detect malignant cells in the blood or bone marrow. However, clinical remission does not necessarily mean that all disease-driving cells have been eliminated. Small amounts of cancer cells may persist below the detection threshold of morphologic examination. This residual disease can later serve as the origin of relapse.
Measurable residual disease (MRD), also referred to as minimal residual disease, describes a very small number of cancer cells that remain during or after treatment and can only be detected by highly sensitive laboratory methods. MRD testing may help assess treatment response, estimate relapse risk, guide further therapy, and support prognosis.
Continuous monitoring of MRD status before clinical relapse can create a diagnostic window in which treatment strategies may still be adapted. This is particularly relevant in hematological malignancies, where MRD has become an important prognostic and monitoring marker, but is also increasingly relevant in solid tumors through ctDNA analysis.
MRD is rarely an obvious, isolated mutation call. It is usually a weak molecular signal embedded in technical noise and biological variability. MRD tracking software must therefore identify mutations, rearrangements, or other tumor-derived markers and evaluate whether these signals provide reproducible evidence of residual disease in a specific patient across multiple time points.
Key terminology
Before we go on, let's define the following terms as they are used in this article:
Measurable/minimal/molecular residual disease (MRD) is the residual tumor burden detectable by molecular methods after treatment.
ctDNA refers to tumor-derived DNA fragments circulating in blood plasma.
Variant Allele Frequency (VAF) is the fraction of mutated molecules (carrying a specific variant) compared to all observed molecules at a certain genomic position. For example, one could say: "In this sample, allele A has a VAF of 10% at chromosome 1, position 5000."
The limit of detection is a measure of analytical sensitivity: it is the lowest ctDNA fraction or variant allele frequency that can be detected with a defined confidence level.
In longitudinal tracking, repeated measurements are taken from the same patient over time.
NGS-based MRD tracking
MRD can be assessed using different technologies, including flow cytometry, karyotyping, fluorescence in situ hybridization (FISH), qPCR, digital PCR, and next-generation sequencing (NGS). This article focuses on NGS-based MRD tracking.
NGS is attractive because it can interrogate many molecular targets simultaneously. Depending on the assay design, the detectable signals may include single nucleotide variants, insertions and deletions, gene rearrangements, copy number alterations, clonotypes, methylation patterns, or fragmentomic features. Furthermore, it is possible to achieve a very high sequencing coverage that allows detecting low-frequency variants with higher confidence, effectively improving the limit of detection. By targeting specific regions, NGS can accurately identify tumor-specific signals and therefore detect MRD with a sensitivity ranging down to 10^-7.
NGS-based MRD analysis creates specific challenges for bioinformatics software. The required sensitivity is often far below that of ordinary somatic variant detection. In routine tumor profiling, a variant may be clinically relevant at a 2-5% variant allele frequency. In MRD tracking, the relevant signal may be present at much lower fractions, close to the intrinsic error rate of sequencing. Therefore, MRD software cannot simply reuse standard variant calling methods and apply a lower threshold.
Tumor-informed and tumor-agnostic MRD assays
Two main conceptual approaches are used in ctDNA-based MRD analysis: tumor-informed and tumor-agnostic assays.
In a tumor-informed assay, the patient's tumor is sequenced first with a larger target region and conventional somatic variant detection methods. This may be done using targeted panels, whole-exome sequencing, or even whole-genome sequencing. From this tumor profile, a set of patient-specific tumor variants is selected. Later samples are then analyzed specifically for these known variants. The software does not need to discover the tumor signals de novo in every follow-up sample. Instead, it can search for predefined molecular signals that are already known to belong to that patient's tumor. This improves analytical sensitivity because the sample preparation and computational methods can focus on a smaller region and a defined set of patient-specific signals.
In a tumor-agnostic assay, plasma is analyzed without prior knowledge of the tumor genome. Such assays may use fixed DNA variant panels, methylation profiles, fragment length patterns, copy number signals, or combinations of molecular markers. The main practical advantage is accessibility. Testing may be possible even when tumor tissue is unavailable, insufficient, or difficult to sequence. The main analytical challenge is that the software must detect a weak tumor-derived signal while suppressing false positives.
What MRD tracking software has to achieve
The clinically useful output of MRD tracking software is an interpretable MRD status, such as MRD positive, MRD negative, or not evaluable. Ideally, this status is accompanied by quantitative evidence, confidence metrics, and assay quality information.
The input for MRD tracking software is the raw sequencing data, sometimes accompanied by a list of variants analyzed with other software. In tumor-informed assays, the software can use a patient-specific list of tumor variants derived from prior tumor sequencing.
The central task is to transform the raw sequencing data into a clinically interpretable molecular signal with very low detection limits. This typically requires several analysis steps, such as quality control, read alignment, molecular barcode processing, ultra-sensitive variant detection, error modeling, longitudinal tracking, and structured reporting.
Analytical challenges
The main challenges in analyzing MRD samples are rooted in the very low abundance of ctDNA or residual malignant molecules after treatment. A sample contains only a limited number of informative DNA molecules that must be reliably captured and carried through sample extraction, library preparation, sequencing, and data analysis. Each of these steps can add technical noise. Variable DNA input, assay-specific artifacts, PCR duplicates, sequencing errors, coverage variability, mapping artifacts, and other sources contribute to background noise and biases that can make it difficult to detect the true biological signals.
For this reason, MRD software often does not simply apply a fixed variant allele frequency threshold. Different genomic loci have different error profiles, and some mutations are more error-prone to detect. Additionally, the coverage varies between targets and between samples. This also means that the software should be adapted to the assay/library preparation.
An important distinction is the difference between "MRD negative" and "not enough information to call MRD negative." A sample with insufficient input DNA, poor coverage, contamination, or index hopping cannot support the same conclusion as a technically successful negative sample. A core requirement of clinical MRD software is, therefore, strict quality control and transparent handling of inconclusive results.
Strategies to improve MRD calling accuracy
Several wet-lab and computational techniques are employed to improve MRD-calling accuracy. Unique molecular identifiers, or UMIs, allow sequencing reads to be grouped by their original DNA molecule. This helps distinguish true molecules from PCR duplicates and enables error suppression through consensus-read generation. By reducing background noise and technical biases, this approach enables MRD detection with significantly improved sensitivity.
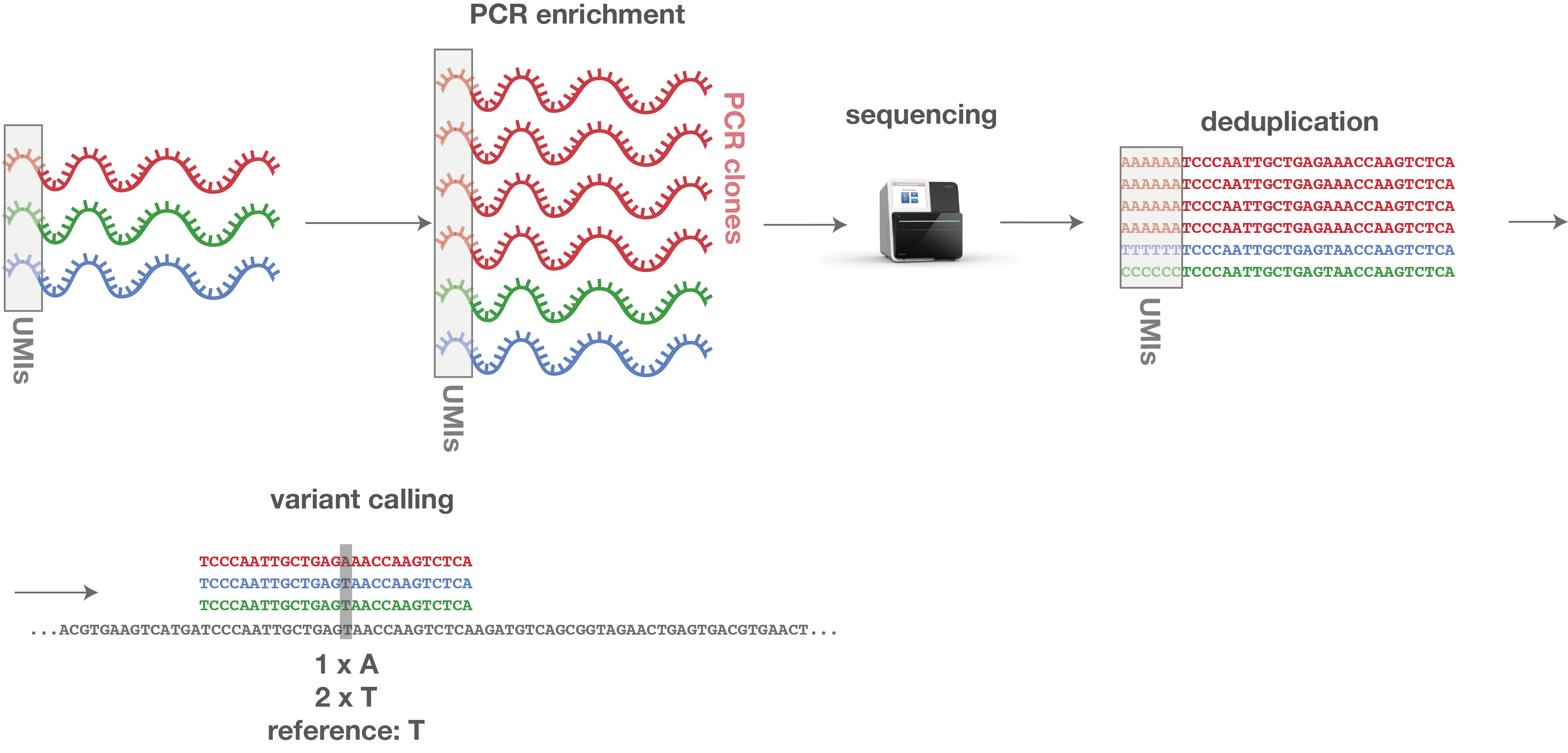 Figure 2: PCR clones can be identified by their same UMI sequence. Thus, they can easily be corrected by deleting all copies, leaving only one representative sequence for each original molecule.
Figure 2: PCR clones can be identified by their same UMI sequence. Thus, they can easily be corrected by deleting all copies, leaving only one representative sequence for each original molecule.
Background error models obtained from reference samples, historical data, or panels of normal samples help identify recurrent technical artifacts. This information can then be used to computationally correct for some of these biases, further improving the accuracy of results.
Quality control should be integrated at multiple checkpoints throughout the workflow, with stringent thresholds applied to input DNA amount, library complexity, coverage uniformity, UMI family size, and contamination.
Notably, error models and quality-control procedures are specific to the assay and sequencing technology used. Therefore, the parameters and modules of any MRD tracking software must be adapted to the laboratory environment; there is no one-size-fits-all data analysis workflow.
Open-source building blocks and commercial software
Unlike general somatic variant detection, MRD tracking is less commonly represented by a single open-source software tool. In principle, a laboratory can assemble an MRD data analysis workflow from established building blocks, including open-source tools for read alignment, UMI processing, variant calling, and quality control, as well as custom software modules for background-error modeling, visualization, and reporting.
Commercial platforms often provide more integrated MRD workflows, particularly when combined with proprietary assays. These systems may include predefined wet-lab protocols, molecular barcode strategies, curated target sets, statistical models, and standardized reports. This simplifies implementation, but it also limits flexibility in adapting assay design or integrating the workflow into local processes.
In tumor-informed assays, the fact that predefined molecular signals are already known from prior sequencing significantly changes the requirements for variant detection. What was previously a trade-off between sensitivity and specificity - discerning true variants from background noise and false positives - is now a task of maximizing sensitivity for known patient-specific signals. Prior knowledge of these signals means that false positives from other genomic regions can largely be neglected. However, this contrasts with how most somatic variant detection tools operate and therefore calls for a customized solution.
General somatic variant analysis software can support parts of an MRD workflow, but it should also be able to handle the specific variant-calling requirements of tumor-informed assays and support the use of UMIs, assay-specific quality control, and error-correction functions. Additional functionality for structured review, storing patient-specific molecular signals for longitudinal analysis, and tracking QC metrics over time is also helpful. Consequently, somatic variant analysis software alone is often not sufficient for MRD tracking.
For commercial tools, bioinformatics workbench software such as Qiagen CLC Genomics Workbench supports the creation of data analysis workflows suitable for MRD tracking. The integrated analysis software Seamless NGS includes specialized ultra-low-frequency variant detection and UMI processing modules for MRD tracking.
Conclusion
MRD tracking software sits at the intersection of molecular diagnostics, longitudinal oncology, and clinical decision support. Its task is different from classical somatic variant detection because it must detect extremely weak molecular signals, suppress technical noise, and interpret the results in the context of patient-specific time courses.
More than in other applications, MRD tracking software is tightly coupled to the assay and sequencing technology used, as the data analysis must be able to support the wet lab techniques such as UMIs or ultra-deep sequencing, but ideally work in close combination with those to reduce errors and biases and achieve the highest possible sensitivity. Reliable MRD tracking thus requires a careful integration of high-coverage sequencing, streamlined sample preparation and laboratory protocols, robust bioinformatics software, and responsible reporting.
Would you like to sharpen your NGS data analysis skills?
Join one of our public workshops!About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on May 28, 2026