Somatic Variant Analysis Software in Molecular Pathology
Today, therapeutic decisions after a cancer diagnosis increasingly depend on the molecular characteristics of the tumor. This is due in part to the availability of targeted therapies such as PARP inhibitors, which are available to selected patients with mutations affecting the BRCA1/2 genes or related DNA repair pathways. Somatic variant detection therefore helps identify tumor-specific DNA changes that may influence prognosis, therapy selection, resistance mechanisms, or eligibility for clinical trials. Because such mutations may be present only in a fraction of cells obtained in a biopsy, reliable detection often requires sensitive sequencing-based approaches. NGS-based variant detection is therefore a central method for identifying clinically relevant mutations, including low-frequency variants that would otherwise be difficult to detect.
Key Terminology
Before we go on, let’s quickly recapitulate three important concepts that we need here.
Somatic mutations are DNA changes that arise in non-reproductive cells during a person’s life. In contrast to germline variants, which are present in reproductive cells and can therefore be found in essentially all cells of the offspring’s body, somatic mutations affect only the descendant cells of the mutated cell. In cancer, such mutations may contribute to tumor initiation, progression, and immune escape.
Variant Allele Frequency (VAF) is the fraction of mutated molecules (carrying a specific variant) compared to all observed molecules at a certain genomic position. For example, one could say: "In this sample, allele A has a VAF of 10% at chromosome 1, position 5000."
Tumor content, or tumor purity, is the fraction of tumor cells in a sample. When a tumor sample is obtained, for example from a breast cancer biopsy, it usually contains a mixture of tumor cells and non-tumor cells. A pathologist can estimate tumor content by visually inspecting the tissue.
💡The lower the tumor content, the more difficult it becomes to detect somatic mutations with a low VAF, because the effective number of DNA sequences that originate from tumor-derived molecules decreases accordingly.
From sequencing data to variant list: Variant detection
The figure below summarizes what sequencing-based variant detection software aims to achieve: given two inputs - the sequencing reads (usually FASTQ or BAM files) and the human reference genome (FASTA file) - we would like to obtain a table of DNA variants (VCF file). The table of DNA variants should include information about the variant's location in the reference (chromosome number and position), the detected genotype (G/T) and additional annotations or statistics that describe the confidence and context of each call.
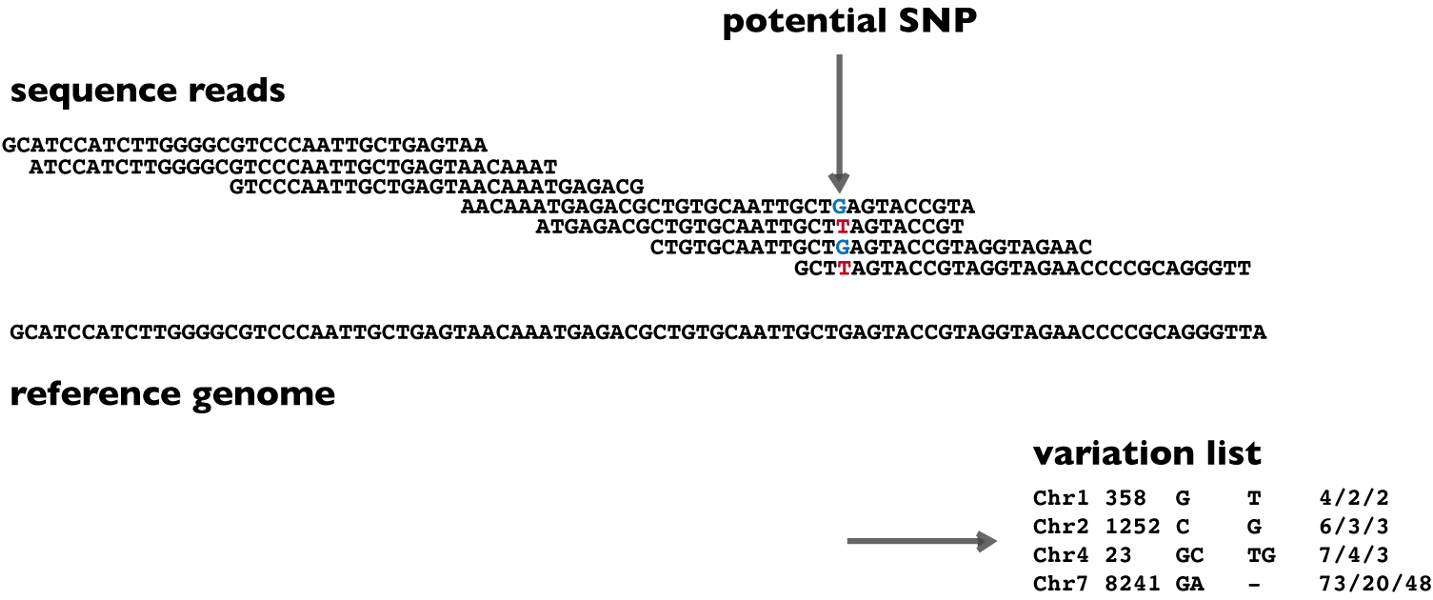
In practice, this task often requires a workflow of several analysis steps. Raw sequencing reads are first subjected to quality control and are then aligned to the reference genome, for example with tools such as BWA-MEM. The resulting alignments are commonly sorted, indexed, and preprocessed. Depending on the application and workflow, this may include duplicate marking, base quality recalibration, local realignment-like procedures, or other normalization steps. Only after this preprocessing does the actual variant calling algorithm evaluate whether the observed read evidence supports a real variant.
In somatic variant detection, this process is more complex than in classical germline variant calling. The caller must account for the fact that the expected allele frequency may be far below 50%, that the sample may contain normal-cell contamination, and that sequencing or sample-preparation artifacts may appear similar to true mutations. Some workflows compare a tumor sample with a matched normal sample from the same patient, while others perform tumor-only analysis and rely more heavily on population databases, panels of normals, and statistical filtering to remove likely germline variants and recurrent artifacts.
Challenges in somatic variant detection
Somatic variant detection is difficult because the signal of interest is often weak, while the amount of technical and biological background noise is substantial. At variant frequencies of 5% or 1%, the difference between a clinically relevant subclonal mutation and a technical artifact may be only a few sequencing reads.
One major challenge is the existence of sequencing errors. Different sequencing platforms and library preparation protocols have characteristic error profiles, and certain sequence contexts are more error-prone than others. PCR amplification can introduce additional artifacts, especially when the same original DNA molecule is amplified many times and the resulting duplicate reads are incorrectly interpreted as independent evidence. This is particularly problematic in targeted panel sequencing, where very high coverage is often used to detect low-frequency variants.
A second challenge is the detection of low-frequency variants themselves. From a statistical perspective, this is a signal-versus-noise problem. The lower the VAF, the more sequencing depth is required to observe enough alternative-supporting molecules. However, increasing the coverage alone does not solve the problem if the additional reads mostly represent amplified copies of the same original molecules or if systematic errors accumulate at the same genomic position. Tumor purity, local copy number, and intratumoral heterogeneity further influence the expected VAF of a true mutation. Low tumor purity and tumor heterogeneity are therefore among the central reasons why clinically relevant variants may appear at low allele frequencies.
A third challenge is sample quality. Many clinical tumor samples are formalin-fixed and paraffin-embedded, which enables long-term storage and routine histopathological processing, but also damages nucleic acids. FFPE processing can fragment DNA and induce chemical modifications, especially cytosine deamination, which can appear as C>T or G>A artifacts in sequencing data.
Finally, alignment errors can produce false-positive or false-negative variant calls. Repetitive regions, homologous genes, pseudogenes, low-complexity sequences, and regions with structural variation are particularly difficult. Reads may be placed at the wrong genomic location or may align with low confidence. In such cases, the variant caller may interpret mapping artifacts as genuine variants, or it may miss real variants because the supporting reads are aligned to the wrong genomic locations.
Strategies to improve accuracy
Several experimental and computational strategies can improve the accuracy of somatic variant detection. Unique Molecular Identifiers (UMIs) are one of the most powerful experimental strategies for reducing technical noise. The principle is to tag DNA molecules with unique sequence barcodes before PCR amplification. Reads that originate from the same original molecule can then be grouped together, and a consensus sequence can be generated. This makes it possible to distinguish errors introduced during PCR or sequencing from variants that were already present in the original DNA molecule.
Advanced variant calling algorithms also play an important role. Modern somatic callers use statistical models, local assembly, Bayesian inference, machine learning, or neural networks to evaluate read evidence. Tumor-normal callers can directly compare the tumor sample with a matched normal sample from the same patient, which helps separate somatic mutations from germline variants. Tumor-only callers, in contrast, are often easier to implement operationally, because no normal sample is required. They, however, need more extensive filtering against population databases, panels of normals, and recurrent technical artifacts.
Filtering and post-processing are therefore central parts of the variant interpretation workflow. A raw variant list usually contains a mixture of true mutations, germline variants, recurrent artifacts, mapping errors, and low-confidence calls. Filtering criteria may include read depth, VAF, base quality, mapping quality, local sequence context, population frequency, and occurrence in a panel of normals. A Panel of Normals is generated from multiple non-tumor samples and helps identify recurrent technical artifacts that appear repeatedly in a specific sequencing workflow.
Overview of open-source variant calling tools
There exists a significant number of open-source or openly available variant detection tools, so it is not possible to compare all of them. The following table gives a subjective selection of important tools that are frequently encountered in somatic variant detection workflows.
| Name | Approach | Description | Release | License | Adoption |
| Mutect2 | Local assembly of haplotypes | Widely used somatic SNV and indel caller from the Broad Institute. It supports tumor-normal and tumor-only workflows and can use resources such as panels of normals and germline databases. | 2018 as part of GATK4 | Apache 2.0 | High |
| VarDict | Heuristic and statistical thresholding | Sensitive variant caller for single-sample and paired analyses. No longer maintained. | 2014 | MIT | Medium |
| Strelka2 | Probabilistic modeling and haplotype-based analysis | Fast small-variant caller for germline and tumor-normal somatic analysis. It remains popular, although the official repository was archived in April 2026. | 2015 | PolyForm Strict 1.0.0 | Medium |
| DeepSomatic | Neural network | DeepVariant-derived somatic caller that uses image-like representations of read evidence and convolutional neural networks. | 2024 | BSD-3-Clause | Emerging |
| VarScan2 | Heuristic and statistical thresholding | Long-established Java-based variant caller for germline, somatic tumor-normal, LOH, and copy-number analyses. | 2010 | Custom. License required for commercial use. | Medium |
What is the best variant calling tool? We could make it simple and say: the best tool is the one that has the greatest adoption, which is most likely Mutect2. However, this would be a gross oversimplification. Before selecting a tool, one should define the relevant comparison criteria, which commonly include:
- High sensitivity for low-frequency variants
- High specificity (few false positives)
- Robustness across different applications, sample types, and coverages
- Support for SNVs, indels, and possibly larger or more complex variants
- Suitability for tumor-normal or tumor-only workflows
- Computational runtime and hardware requirements
- Availability of documentation and long-term maintenance
- License and suitability for academic, clinical or commercial use
The developers report much of this information in their respective publications and documentation. However, when it comes to comparisons with other tools, independent benchmarks are often more informative. New benchmarks for somatic variant callers are published regularly, which is appropriate because tools, tool versions, sequencing chemistries, and reference datasets continue to change.
Examples from 2025 include a benchmark of 20 somatic variant callers for whole-exome sequencing, which found that different callers performed best for different variant classes. Another benchmark focused on mutation calling in circulating tumor DNA, where low VAFs and DNA degradation make the problem even more difficult.
However, we do not recommend looking only at the most recent benchmark and selecting whatever tool comes out on top. Benchmarks are highly dependent on the data type, truth set, variant classes, filtering settings, and performance metric. In many real-world applications, “soft” criteria such as interoperability, documentation quality, community adoption, and long-term support are equally important. A tool that performs slightly better in one benchmark may still be less suitable for a clinical laboratory if it is difficult to integrate, poorly documented, or insufficiently maintained.
Commercial integrated solutions
Let us now look at selected commercial offerings, focusing on clinical applications in precision medicine. The main difference compared with the open-source tools discussed above is that commercial platforms are usually not limited to the specific task of somatic variant detection. Instead, they are often end-to-end genomic analysis platforms that cover several steps of the clinical workflow:
- Integrated pipelines from raw data to reporting, including alignment, variant calling, annotation, filtering
- User-friendly interfaces designed for clinical laboratories
- Tools for variant interpretation, classification, and report generation
- Integrated databases for samples, variants, quality control, and previous interpretations
- Knowledge bases for clinical evidence, drugs, guidelines, and literature
- Workflow automation to reduce manual steps and turnaround time
- Runtime optimizations and operational robustness
- User and role management for laboratory teams
- Audit trails, versioning, and regulatory compliance features, if applicable
| Name | Available since | Description |
| SOPHiA GENETICS | 2011 | Cloud-based, AI-supported analytics platform for precision medicine. |
| QIAGEN QCI Interpret | 2018 (in this context) | Clinical decision-support and variant interpretation platform. |
| Pierian | 2014 | Clinical genomics workflow platform for variant interpretation, classification, and reporting for Illumina TruSight Oncology 500 |
| Seamless NGS | 2018 | Local-deployable platform for automated NGS data analysis and management. |
Conclusion
Somatic variant detection is a technically demanding but clinically important component of precision oncology. Its purpose is to identify tumor-derived mutations that may influence diagnosis, prognosis, therapy selection, or clinical trial eligibility. This requires reliable detection of variants that may occur at low allele frequencies. The main challenges include sequencing errors, PCR artifacts, low tumor purity, limited coverage, FFPE-induced damage, and alignment ambiguities. These challenges can be addressed by combining appropriate laboratory methods, such as UMIs and sufficient molecular input, with robust computational strategies, such as advanced variant callers, tumor-normal comparison, panels of normals, artifact filtering, and careful post-processing. Continued innovation in sequencing methods, sample preparation, and clinical interpretation will further improve the field.
Would you like to sharpen your NGS data analysis skills?
Join one of our public workshops!About us
ecSeq is a bioinformatics solution provider with solid expertise in the analysis of high-throughput sequencing data. We can help you to get the most out of your sequencing experiments by developing data analysis strategies and expert consulting. We organize public workshops and conduct on-site trainings on NGS data analysis.
Last updated on April 30, 2026